WhichTF is dominant in your open chromatin data?
Authors
Y Tanigawa, ES Dyer, G Bejerano
Abstract
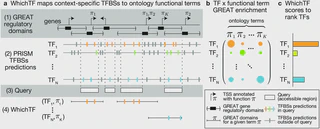
We present WhichTF, a computational method to identify functionally important transcription factors (TFs) from chromatin accessibility measurements. To rank TFs, WhichTF applies an ontology-guided functional approach to compute novel enrichment by integrating accessibility measurements, high-confidence pre-computed conservation-aware TF binding sites, and putative gene-regulatory models.
We present WhichTF, a computational method to identify functionally important
transcription factors (TFs) from chromatin accessibility measurements. To rank TFs,
WhichTF applies an ontology-guided functional approach to compute novel enrichment by
integrating accessibility measurements, high-confidence pre-computed conservation-aware
TF binding sites, and putative gene-regulatory models. Comparison with prior sheer
abundance-based methods reveals the unique ability of WhichTF to identify
context-specific TFs with functional relevance, including NF-κB family members in
lymphocytes and GATA factors in cardiac cells. To distinguish the transcriptional
regulatory landscape in closely related samples, we apply differential analysis and
demonstrate its utility in lymphocyte, mesoderm developmental, and disease cells. We
find suggestive, under-characterized TFs, such as RUNX3 in mesoderm development and GLI1
in systemic lupus erythematosus. We also find TFs known for stress response, suggesting
routine experimental caveats that warrant careful consideration. WhichTF yields
biological insight into known and novel molecular mechanisms of TF-mediated
transcriptional regulation in diverse contexts, including human and mouse cell types,
cell fate trajectories, and disease-associated cells.
Type
Publication
Published in PLOS Comput Biol, 2022
We develop an ontology-guided approach to ranking tissue-/cell-type-specific transcription factors (TFs) from chromatin accessibility data.