A cross-population atlas of genetic associations for 220 human phenotypes
Sep 30, 2021
·
1 min read
Authors
S Sakaue, M Kanai, Y Tanigawa, J Karjalainen, M Kurki, S Koshiba, A Narita, T Konuma, K Yamamoto, M Akiyama, K Ishigaki, A Suzuki, K Suzuki, W Obara, K Yamaji, K Takahashi, S Asai, Y Takahashi, T Suzuki, N Shinozaki, H Yamaguchi, S Minami, S Murayama, K Yoshimori, S Nagayama, D Obata, M Higashiyama, A Masumoto, Y Koretsune, FinnGen, K Ito, C Terao, T Yamauchi, I Komuro, T Kadowaki, G Tamiya, M Yamamoto, Y Nakamura, M Kubo, Y Murakami, K Yamamoto, Y Kamatani, A Palotie, MA Rivas, MJ Daly, K Matsuda, Y Okada
Abstract
Current genome-wide association studies do not yet capture sufficient diversity in populations and scope of phenotypes. To expand an atlas of genetic associations in non-European populations, we conducted 220 deep-phenotype genome-wide association studies (diseases, biomarkers and medication usage) in BioBank Japan (n = 179,000), by incorporating past medical history and text-mining of electronic medical records.
Current genome-wide association studies do not yet capture sufficient diversity in
populations and scope of phenotypes. To expand an atlas of genetic associations in
non-European populations, we conducted 220 deep-phenotype genome-wide association
studies (diseases, biomarkers and medication usage) in BioBank Japan (n = 179,000), by
incorporating past medical history and text-mining of electronic medical records.
Meta-analyses with the UK Biobank and FinnGen (ntotal = 628,000) identified ~5,000 new
loci, which improved the resolution of the genomic map of human traits. This atlas
elucidated the landscape of pleiotropy as represented by the major histocompatibility
complex locus, where we conducted HLA fine-mapping. Finally, we performed statistical
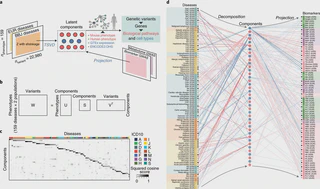
decomposition of matrices of phenome-wide summary statistics, and identified latent
genetic components, which pinpointed responsible variants and biological mechanisms
underlying current disease classifications across populations. The decomposed
components enabled genetically informed subtyping of similar diseases (for example,
allergic diseases). Our study suggests a potential avenue for hypothesis-free
re-investigation of human diseases through genetics.
Type
Publication
Published in Nature Genetics, 2021
Using a set of GWAS summary statistics of diseases characterized from both European (UK Biobank and FinnGen) and East Asian (Biobank Japan) populations, we dissected latent DeGAs components of multi-ethnic association summary statistics.